With the rising number of complex biologics currently in translation research, exploratory, and proof of concept (POC) studies, there is growing pressure for drug developers to reduce project timelines to Investigational New Drug (IND) submission. This requires a holistic approach to biologic drug development to advance from the gene sequence to the generation of the drug substance and drug product as quickly as possible. This will also demand the coordination of key overlapping activities and a paradigm shift in targeted cancer diagnosis and therapeutics to meet the appropriate IND-enabling activities.
Starting with the gene sequence, a robust cell line development (CLD) strategy is the foundation of this holistic approach with consideration of methodologies that minimize delays and associated risks. Traditionally, first-in-human phase I and toxicology material are generated using clonal-derived cells. However, the use of stable cell pools as alternative non-clonal platforms brings the opportunity to produce biologics faster, as well as more cost-effectively, with quality and yields similar to clonal cell lines.
First-in-human phase 0 studies (also known as microdosing POC studies) are designed to speed up the development of promising drugs or imaging agents, by establishing very early on whether the drug or agent behaves in human subjects as expected from preclinical studies. Distinctive features of phase 0 trials include the administration of single subtherapeutic doses of the study drug to typically a few (≤5) subjects. This trial gathers preliminary human in vivo data on pharmacokinetics (how the body processes the drug), pharmacodynamics (how the drug works in the body) and target (i.e., the interaction between the drug and the target protein or gene to obtain a biological response) much sooner for early to late clinical human trials. They enable go/no-go decisions to be based on relevant human models instead of relying on sometimes inconsistent animal data. A phase 0 study gives no data on safety or efficacy, being by definition a dose too low (typically ≤100µg) to cause any therapeutic effect and less risk of human toxicity.1
In this article, we propose an accelerated and experience-supported approach with a 5-month timeline to produce radioimmune conjugate pharmaceuticals for first-in-human phase 0 POC studies starting from stable pool cells to generate the harvest cell culture fluid (HCCF).

The Market Potential of Theranostics (and Phase 0 Application)
Once considered niche cancer therapy, radioligands are in a renaissance. Theranostics technology pertains to the generation of radioimmune conjugates that can be used in both diagnosis (molecular imaging) and therapeutics. They have demonstrated significant promise for complementing and supplementing immunotherapy as a personalized cancer treatment. The advent of the phase 0 POC imaging human study has become a precursor to theranostics for phase Ia (imaging) and Ib (therapy) human clinical studies. The value of the global theranostics market is expected to maintain a compound annual growth rate (CAGR) of ~9.5–12.2% in the coming years, reaching $124–$153 billion by 2028.2 This is exemplified by the recent U.S. approvals of Luthathera and Pluvicto (both Lu-177–based radionuclides) in 2018 and 2022, respectively, by Novartis, anticipating that Pluvicto is expected to exceed $2 billion in peak sales. To this end, Novartis’ big pharma peers, such as Eli Lilly and Bristol Myers Squibb, both announced buyouts of radioligand therapy companies during the past year in the billions of dollars.
Faster Pathway to Phase 0 POC Human Studies of Radiopharmaceutical Drugs for Diagnosis
The traditional method to identify suitable manufacturing cell lines comprises time-consuming CLD, where typically thousands of stable mAb expressing clones are generated and screened to achieve the research cell bank (RCB) in about 6 months. Therefore, 15 to 24 months are needed for established drug development concepts, spanning from the start of cell line generation to the start of a first-In-human phase I clinical evaluation of the antibody–chelator conjugate.3 Alternatively, the use of stable cell line pools has great potential to significantly accelerate the timeline for biopharmaceutical development. However, regulatory authorities had not accepted the use of stable pools rather than clonal cell lines until recently, and its use now includes the production of material for toxicological and preclinical studies. In addition, with recent and significant improvements in performance, stable pools are now capable of producing recombinant proteins more rapidly and cost-effectively with yields and quality similar to clonal cell lines.4
Recently, the U.S. Food and Drug Administration (FDA) published guidance for the industry following the nonclinical studies recommendations for the diagnosis of microdose radiopharmaceuticals and the current International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) M3. The guidance provides the regulatory framework for the application of phase 0 approaches called “exploratory clinical trials.”5,6,7 The FDA recommends the administration of a microdose of ≤100 µg of the drug in human studies, which is the lower end of the dose–response curve. Also, dose-related adverse events are unlikely to occur, allowing safety concerns not to be a problem.5 Microdosing of radiopharmaceuticals for diagnostic imaging is part of phase 0 drug development, providing the opportunity to directly test the drug in humans and obtain more accurate pharmacokinetics and pharmacodynamics data than animal studies. The FDA advises phase 0 scheduling for new radiopharmaceutical diagnostic drugs to facilitate the timely conduct of clinical trials by providing information that supports a more effective design for phases I and II. This also decreases unnecessary practice with animals and other resources for the trials. Phase 0 methodologies have the capability to improve preclinical candidate selection and enable safer, cheaper, quicker (typically 1 month or less), and more informed developmental decisions in most drug development scenarios.7,8 To this end and as a precursor to theranostics, radioimmune conjugates used in phase 0 human studies will accelerate the development of theranostics. This has now been established for use in both diagnostics and therapeutics of cancer treatment or other modalities from early-phase clinical trials through to late phase and commercial launch.8
Process Development and GMP Manufacturing of the mAb and mAb–Chelator Conjugate
It should be noted that the process development and GMP manufacturing processes performed for the generation of the purified “naked” mAb and the corresponding mAb–chelator conjugate proposed in this article are based on GBI’s robust platform processes borne from over 20 years of experience with these products. Minimal optimization will be required for these activities depending specifically on the project.2,9
Process Development (PD) of the mAb
Upstream (Cell Culture and Bioreactor) Manufacturing of the mAb
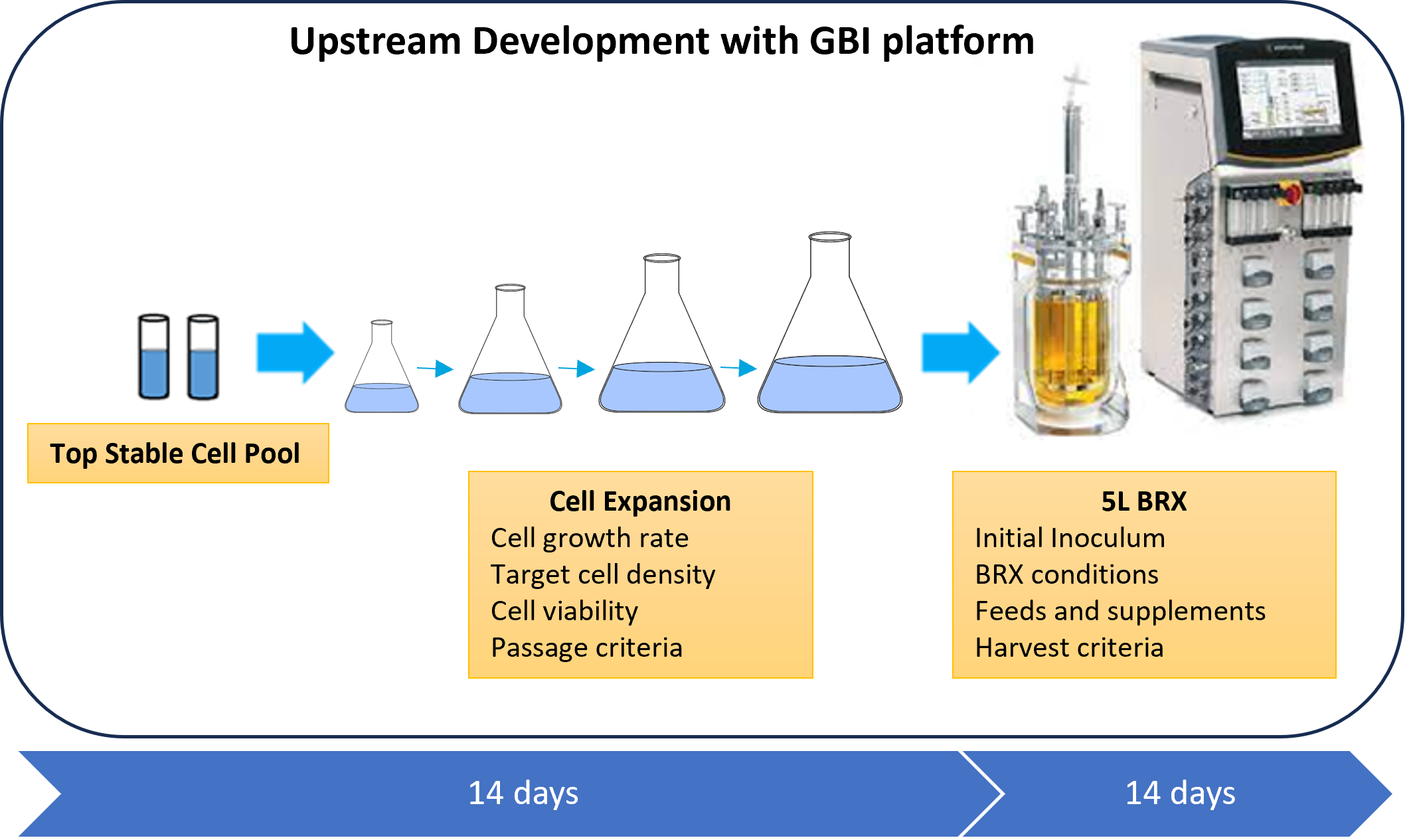
Starting with the frozen stable pool bank (with an expected titer of ≥ 1g/L of mAb in the harvest cell culture fluid (HCCF) with a CHO cell line), one production run is typically performed in the process development lab, applying a very well-characterized and established upstream platform from over 100 projects performed at GBI to obtain the HCCF. The upstream development consists of thawing a vial of the stable cell pool bank and expanding the cells until enough cells are generated to inoculate a 5-L bioreactor. This allows us to evaluate and confirm the expansion parameters, such as viable cell seeding density, cell culture conditions, passage criteria, and number of passages. The stable pool growth rate and viability of the cells will also be defined. A 5-L scale bioreactor run is usually sufficient to produce the monoclonal antibody needed and allows evaluation of the bioreactor performance in terms of cell growth, protein production, and product quality attributes. Bioreactor process parameters, such as initial inoculum, dissolved oxygen, agitation, temperature shift, feeds, and supplements, will be defined. All this information is crucial to performing the GMP manufacturing run. The cell culture and bioreactor production time to generate the HCCF is expected to take about 4 weeks to complete (see Figure 1).
Figure 1: Upstream process timeline from the stable pool cell bank through production of the harvest cell culture fluid (HCCF).
Downstream Purification and Characterization of the mAb
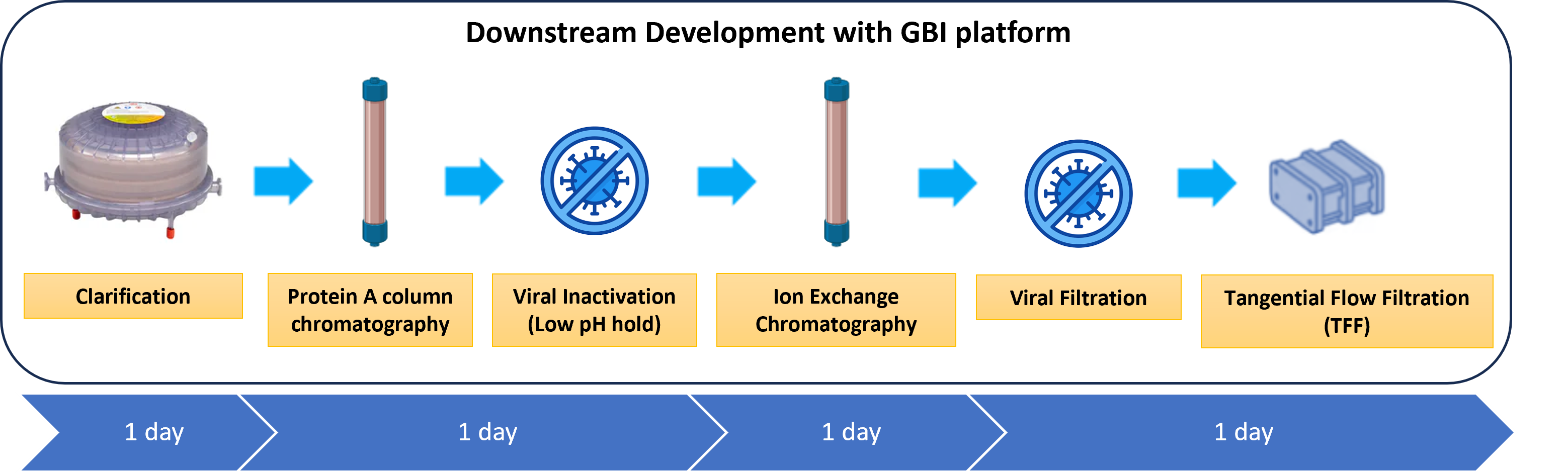
GBI will adapt its downstream platform to perform robust mAb purification in order to achieve the required product yield, purity, and quality specifications. The downstream stage will allow us to identify the impact of the most important parameters on the product quality attributes and clearance of impurities across the purification process. The HCCF generated during upstream PD will be clarified and purified. For this run, a two-column purification and two orthogonal viral clearance steps will be performed in the following unit operations (See Figure 2):
- HCCF clarification
- Capture step by Protein A column chromatography to remove host cell contaminants, including proteins, DNA, endotoxins, and viruses. Static binding capacity will be determined.
- Viral inactivation step of enveloped viruses (low pH hold at 3.5 for 1 hour).
- Polishing step chromatography (bind and elute) to remove high-molecular-weight (HMW) and low-molecular-weight (LMW) variants of the product, host cell contaminants, and leached Protein A, among others.
- Viral filtration step removal of viral contaminants (≥ 20 nm)
- Tangential flow filtration for concentration and diafiltration of the mAb into an appropriate buffer formulation
Figure 2: Downstream purification process timeline.
Characterization of the purified mAb for product quality and safety will be performed and will include appearance, A280, pH, residual HCP, SEC-HPLC, residual Pro A, icIEF, CE-SDS (R/NR), antigen-binding ELISA, residual DNA, and endotoxin.9 Other optional extended characterization, such as N-glycan profile, peptide map, and intact mass, may also be performed.
GMP Manufacturing of the mAb
Upon completion of a single run of the upstream and downstream process development activities of the mAb, a GMP manufacturing run will be performed using a manufacturing batch record (MBR) generated from the development run at a 5-L bioreactor scale and conditions similar to the PD run. A GMP master cell bank (MCB) will be created upon receipt of the stable pool cell bank, in parallel with the upstream PD activities and before the GMP manufacturing run in a GMP core facility. For the subsequent process development and GMP manufacturing of the corresponding mAb–chelator conjugate, approximately 1–2 grams of the purified antibody produced in the GMP run should suffice, since only ≤100 µg dosing of the conjugate into a few patients is required for phase 0 human studies.
Bioconjugation of Purified mAb with Appropriate Chelator: Process Development and GMP Manufacturing
Types of Radionuclides Used – Alpha versus Beta versus Gamma emitters
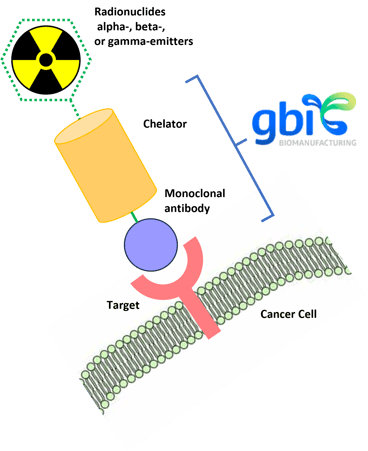
Radioimmune-conjugated pharmaceuticals (radiolabeled antibodies)10 used as payloads in phase 0 and theranostic applications are either alpha-, beta-, or gamma-emitters (see Figure 3). Beta-emitters deliver high-energy electrons that can travel about two to six millimeters and work well in targets with heterogeneous expression systems. These particles tend to cause single-stranded DNA breaks, which can be effective in stopping tumor progression and triggering cell death. Some common beta emitter radionuclides used in imaging and therapy include yttrium-90, lutetium-177, and iodine-131. Alpha particles are the nuclei of helium atoms (two protons and two neutrons). Alpha particles, such as those emitted by actinium-225, bismuth-213 and lead-212, can deliver much larger emissions (the size of a nucleus versus an electron) and are generally more potent, causing double-stranded DNA breaks, compared with beta particles. These particles only travel around one to three cells, so they are in a more selective range but can cause more damage to the cancer cells while reducing their effects on healthy tissues. Alpha-emitters can be blocked by a piece of paper, which makes them easier to shield and the drug supply chain distribution and handling process more convenient. It should also be noted that alpha-emitters can be used to treat patients who have not responded to beta-emitters. Gamma particles are photons of high-energy electromagnetic radiation that include certain isotopes, such as iodine.
Figure 3: Radioimmune-conjugated pharmaceutical scheme.
Types of Chelators Used in the Production of Radioimmune Conjugates
Chelators play a crucial role in the development of radioimmune conjugates by binding the radioactive isotopes and linking them to targeting molecules, such as antibodies or peptides. These chelators vary in their properties, such as stability, affinity for specific radiometals, and ease of conjugation, thus allowing researchers to select the most appropriate chelators based upon the specific requirements of the radioimmune conjugates. To this end, for diagnostic applications, the radionuclide must remain bound to the chelator, while for therapeutics, the chelator must release the payload at the target site. To date, there have only been a limited number of commercially available chelators for use in theranostic applications, including cyclic 1,4,7,10-Tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA), [(ʀ)-2-Amino-3-(4-isothiocyanatophenyl)propyl]-trans-(S,S)-cyclohexane-1,2-diamine-pentaacetic acid (acyclic “CHX-A” linkers), TCMC (also known as DOTAM), and deferoxamine (DFO). DOTA and DFO are commonly used to produce diagnostic products, while CHX-A is used for therapeutics. Ongoing research aims to develop new chelators with improved characteristics for enhanced performance in radioimmune conjugates.
PD and GMP Manufacturing of the mAb-Chelator Conjugate
With regards to the PD production of the mAb–chelator conjugate, GBI will utilize its conjugation platform to run the process once at up to 1-g scale and characterize. A limited buffer stability (up to 3 months) of the conjugate prepared (in the PD lab) will be performed in parallel to the start of the GMP production of the conjugate (in the GMP facility) at a similar scale and conditions performed for the PD run. The PD and GMP mAb–chelator conjugates will be manufactured from the respective mAbs generated from the PD and GMP purification runs described above. They will be tested for product quality and safety via appearance, A280, pH, SEC-HPLC, icIEF, CE-SDS (R/NR), antigen-binding ELISA, chelator:antibody mole ratio (CAR), endotoxin, and bioburden.2,9 It should be noted that safety is not a requirement as per the FDA guidance.5 However, it is highly recommended that a robust conjugation development process be performed, which ensures all critical quality attributes and safety requirements of the mAb and the mAb–chelator conjugate are achieved.
GBI has been developing and manufacturing a wide range of bioconjugates for more than two decades. Many projects have involved radioisotopes, fluorescent dyes, and other payloads that are suitable for theranostic applications (Table 1).
Table 1: Selected Radioimmune Conjugates Manufactured by GBI
| GBI Code | Protein : Payload | Project Status |
| Antibody–Chelator Conjugates | ||
| 379 | IgG Monoclonal Antibody: DOTA Chelator (In111 Isotope) | PD |
| 378 | IgG Monoclonal Antibody: CHX-A” Chelator (In111 Isotope) | GMP MFG* |
| 377 | IgG Monoclonal Antibody : CHX-A” Chelator (Y90 Isotope) | GMP* |
| 374 | IgG Monoclonal Antibody : Direct Conjugation of I131 Isotope | GMP* (Phase III; multiple batches; BLA-enabling activities) |
| 363 | Recombinant Fusion Protein : DOTA Chelator (Cu64 Isotope) | GMP* |
| 353 | IgG Monoclonal Antibody : CHX-A” Chelator (In111 Isotope) | GMP* |
| 346 | IgG Monoclonal Antibody : CHX-A” Chelator (Bi213 Isotope) | GMP* |
| 345 | IgG Monoclonal Antibody : CHX-A” Chelator (In111 Isotope) | GMP* |
| 340 | IgG Monoclonal Antibody : CHX-A” Chelator (Bi213 Isotope) | Tox |
| 339 | IgG Monoclonal Antibody : TCMC Chelator (Pb212 Isotope) | GMP* |
| 320 | IgG Monoclonal Antibody : CHX-A” Chelator (In111 Isotope) | GMP* |
| 312 | IgM Monoclonal Antibody : Direct Conjugation of Re288 Isotope | Multiple GMP batches* |
| 298 | IgG Monoclonal Antibody : CHX-A” Chelator (In111 Isotope) | GMP* |
| 243 | IgG Monoclonal Antibody: DOTA Chelator (Lu177 Isotope) | GMP* |
| 384 | IgG Monoclonal Antibodies : DOTA Chelator (Lu177 Isotope) | GMP* |
| 415 | Murine Antibody : DOTA Chelator (Lu177 Isotope) | Conformance |
| 428 | IgG Monoclonal Antibody : Direct Conjugation of Ac225 Isotope | GMP* |
| 434 | IgG Monoclonal Antibody: DFO TPP Chelator (Zr89 Isotope) | GMP*(Phase III; multiple batches; BLA-enabling activities) |
| 436 | IgG Monoclonal Antibody: CHX-A’ (Bi111) | PD |
| 510 | IgG Monoclonal Antibody: DFO TPP Chelator (Zr89 Isotope) | PPQ runs (3) for commercial launch |
*Denotes projects including process development (PD), scale-up, and cGMP manufacturing
Proposed timelines to produce monoclonal antibody conjugated with chelator for phase 0 and phase I
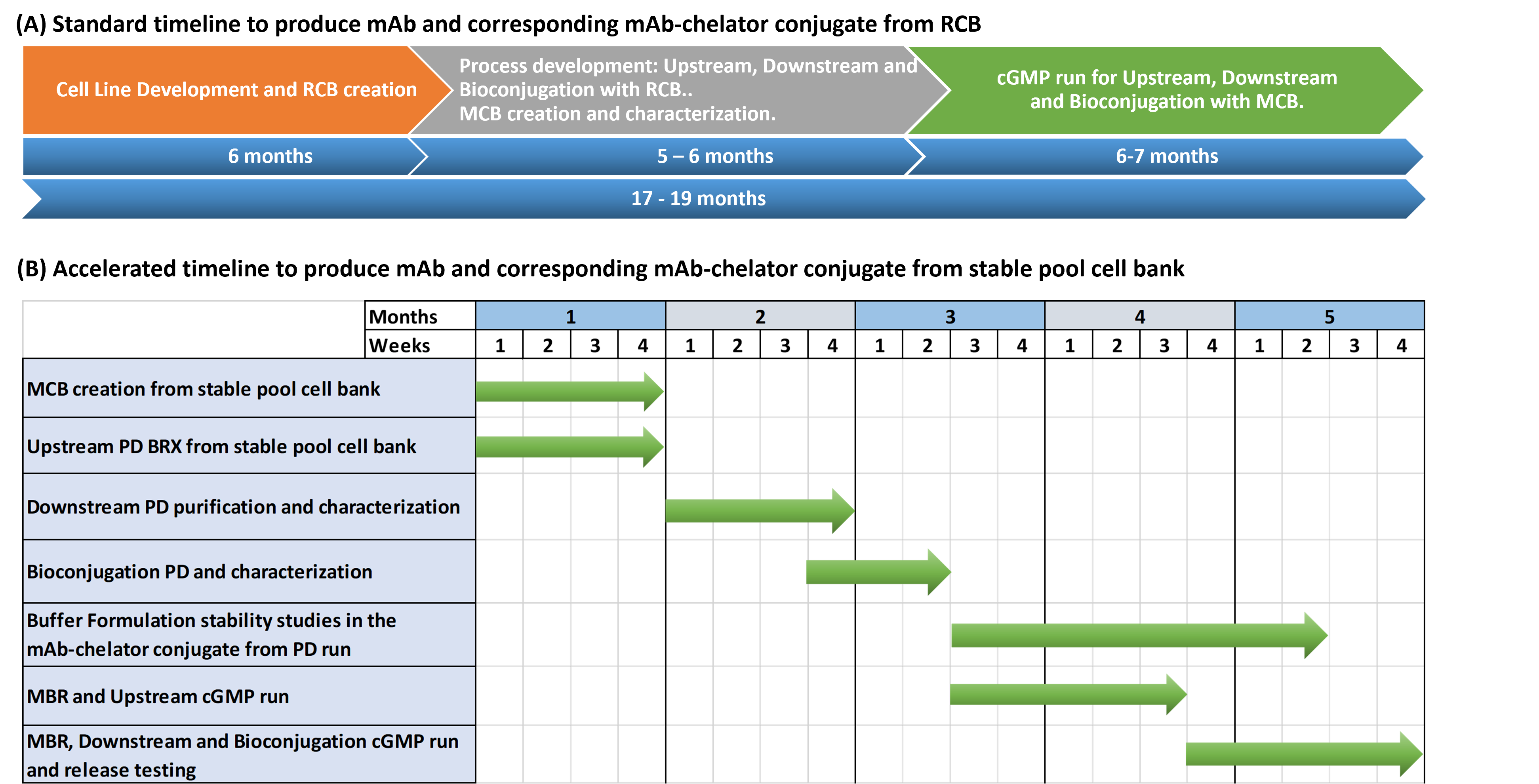
Figure 4: Timelines to produce mAb–chelator conjugate
Figure 4 (A) shows the standard timeline starting from RCB creation by a CLD company and transferred to GBI. Activities involve MCB creation and characterization, process development, and cGMP manufacturing with scaling to produce the mAb drug substance and the corresponding mAb–chelator conjugate drug substance and drug product to be used for early-phase clinical trials. The total timeline is about 17–19 months, with GBI’s activities requiring about 11 to 13 months.9
Figure 4 (B) describes the accelerated timeline, starting from a stable cell pool bank (from the client) through process development, MCB creation from a stable pool cell bank, and cGMP manufacturing to produce the mAb and corresponding mA–chelator conjugate drug substance and drug product to be used for phase 0 POC human studies. The timeline for the production of the conjugate from the stable pool cell bank is proposed as 5 months.
Conclusion
With the recent advancements and the enormous potential of theranostics technology as it pertains to radioimmune conjugates for personalized diagnosis and treatment of cancer, the utilization of radiopharmaceutical drugs for phase 0 human studies (as a precursor to theranostics) is poised to become a standard and critical regulatory event toward rapid and cost-saving pharmaceutical drug development and manufacturing during the next few years and beyond. GBI Biomanufacturing is a U.S.-based contract development and manufacturing organization (CDMO) with over 20 years of experience, innovation, and expertise in the radioimmune conjugates (and many other bioconjugates) arena. During this time, GBI has partnered with many clients and successfully developed scalable, compliant, and economic platforms for all facets of process development, GMP manufacturing, and IND/BLA-enabling activities of “naked” mAbs (and many other complex biologics) and the corresponding mAb–chelator conjugates for the generation of radiopharmaceuticals. This will provide expedited manufacturing solutions to support phase 0 and facilitate the advancement to phase Ia/Ib clinical trials with theranostics applications.
References:
- Sesay, M. “What do you see as the most significant regulatory decision or guidance on the horizon for 2024 (or beyond)?” Pharma’s Almanac. 15 Dec. 2023
- Sesay, M. “Theranostic Manufacturing Solutions.” Pharma’s Almanac. 29 Aug. 2022
- Schmieder, V., Fieder, J., Drerup, R., Gutierrez, E. A., Guelch, C., Stolzenberger, J., & Fischer, S. “Towards maximum acceleration of monoclonal antibody development: Leveraging transposase-mediated cell line generation to enable GMP manufacturing within 3 months using a stable pool.” Journal of Biotechnology. 349: 53–64 (2022).
- Joubert, S., Stuible, M., Lord‐Dufour, S., Lamoureux, L., Vaillancourt, F., Perret, S., & Durocher, Y. “A CHO stable pool production platform for rapid clinical development of trimeric SARS‐CoV‐2 spike subunit vaccine antigens ” Biotechnology and Bioengineering 120: 1746–1761 (2023).
- “Microdose Radiopharmaceutical Diagnostic Drugs: Nonclinical Study Recommendations.” U.S. Food and Drug Administration. 2018.
”Guidance on Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals M3(R2) 8–16 ” International Council for Harmonization Secretariat. 2009.
- Burt, T., Roffel, A. F., Langer, O., Anderson, K., & DiMasi, J. “Strategic, feasibility, economic, and cultural aspects of phase 0 approaches: Is it time to change the drug development process in order to increase productivity?” Clinical and Translational Science 15: 1355–1379 (2022).
- Burt, T., Young, G., Lee, W., Kusuhara, H., Langer, O., Rowland, M., & Sugiyama, Y. “Phase 0/microdosing approaches: time for mainstream application in drug development?” Nature Reviews Drug Discovery. 19: 801–818 (2020).
- Sesay, M and Majdoch, A.“Leveraging Experience to Reduce Timelines for IND-Enabling Activities.” Pharma’s Almanac. 19 Sep. 2022
- Parakh, S., Lee, S. T., Gan, H. K., & Scott, A. M. “Radiolabeled antibodies for cancer imaging and therapy”. Cancers. 14: 1454 (2022).